1 Toolkit for network analysis
Alessandro Vindigni
session 16/03/2023
An ecological network is a dataset describing a group of species and their reciprocal interaction. Typically, this data is collected by ecologists in the field, even if for some purposes ecological networks can also be generated by computer simulations. In recent publications experimental datasets are usually made available, either as supplemental material or through a repository on the web. Some research groups have also gathered network datasets from different publications into public databases to facilitate access to the scientific community. Two of such examples are Mangal and Web of life.
In this session you will learn how to download datasets from the Web of life database of experimental ecological networks and convert them into several formats that are suitable for analysis. Finally, we will compute one network property – the connectance – and plot it.
1.1 The web of life
Web of life is a database of ecological networks developed and maintained by the Bascompte lab. More information about the project can be found in this paper, while here you can find a manual to download ecological networks manually. In this section you will learn to automate downloading the networks available on web of life, visualize them, and perform some basic calculation using those data.
This web interface provides a visualization of where data of a given network have been collected over the globe.
Clicking the dot corresponding to a network, the relative metadata can be visualized as well as a 3D representation of the network.
The incidence matrix is also displayed and can be downloaded in different formats.
Networks are named according to the following convention
- interaction type: mutualistic (M) or antagonistic (A)
- network type: Plant-Ant (PAO), Plant-Epiphyte (PE), Pollination (PL), Seed Dispersal (SD), Food Webs (FW), Host-Parasite (HP), Host-Parasitoid (HPD), Plant-Herbivore (PH), Anemone-Fish (AF)
- id labeling a specific network.
1.1.1 Download one network from the weboflife API
In a research projects it is often convenient to bypass the web interface and store data directly in a dataframe using a command line. This has several advantages in terms of efficiency and reproducibility of the analysis you are conducting. We will see how to do this in R using the function fromJSON() of the package jsonlite, but similar commands can be run in python or in other languages. To this aim, we have opened few dedicated endpoints on the Application Programming Interface (API) of the web-of-life project.
Let us focus on the network number 002 of the Seed Dispersal type, named M_SD_002: the set of commands to download it are
# define the base url
base_url <- "https://www.web-of-life.es/"
# define the full url associated with the endpoint that download networks interactions
json_url <- paste0(base_url,"get_networks.php?network_name=M_SD_002")
# import the jsonlite package (if not done at the beginning)
library(rjson)
# and run it
SD_002_nw <- jsonlite::fromJSON(json_url)It is always good practice to have a look at the class of a newly defined object (dataframe) and display its first rows to understand how this data looks like, number of columns, their names, etc.
# check the class
class(SD_002_nw)## [1] "data.frame"# have a look at the dataframe
head(SD_002_nw)## network_name species1 species2
## 1 M_SD_002 Manucodia keraudrenii Cissus hypoglauca
## 2 M_SD_002 Manucodia keraudrenii Ficus gul
## 3 M_SD_002 Manucodia keraudrenii Ficus odoardi
## 4 M_SD_002 Manucodia keraudrenii Chisocheton sp1 M_SD_002
## 5 M_SD_002 Manucodia keraudrenii Dysoxylum sp1 M_SD_002
## 6 M_SD_002 Manucodia keraudrenii Elmerrillia sp1 M_SD_002
## connection_strength
## 1 13
## 2 155
## 3 55
## 4 7
## 5 1
## 6 8The content of a dataframe can be visualized more nicely using the package formattable
# import the dplyr package (if not done at the beginning)
library(formattable)
# visualize the dataframe in nicer way
formattable(head(SD_002_nw)) # visualize the dataframe in anicer way| network_name | species1 | species2 | connection_strength |
|---|---|---|---|
| M_SD_002 | Manucodia keraudrenii | Cissus hypoglauca | 13 |
| M_SD_002 | Manucodia keraudrenii | Ficus gul | 155 |
| M_SD_002 | Manucodia keraudrenii | Ficus odoardi | 55 |
| M_SD_002 | Manucodia keraudrenii | Chisocheton sp1 M_SD_002 | 7 |
| M_SD_002 | Manucodia keraudrenii | Dysoxylum sp1 M_SD_002 | 1 |
| M_SD_002 | Manucodia keraudrenii | Elmerrillia sp1 M_SD_002 | 8 |
# formattable(SD_002_nw) # to see all the rows1.1.2 Download multiple networks from the weboflife API
If you hit the endpoint https://www.web-of-life.es/get_networks.php without passing any argument into the url, all the networks available in the database are returned, namely a dataframe with columns
network_name, species1, species2, connection_strength:
json_url <- paste0(base_url,"get_networks.php")
all_nws <- jsonlite::fromJSON(json_url)
# show results
formattable(head(all_nws))| network_name | species1 | species2 | connection_strength |
|---|---|---|---|
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus euxinicus | 3 |
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus hypanis | 3 |
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus proximus | 74 |
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus shovi | 13 |
| A_HP_001 | Apodemus mystacinus | Hystrichopsylla satunini | 4 |
| A_HP_001 | Apodemus mystacinus | Leptopsylla segnis | 11 |
You can list and count all the distinct networks you have downloaded using the function distinct() of the dplyr package for data manipulation:
# import the formattable package (if not done at the beginning)
library(dplyr)
all_nw_names <- distinct(all_nws, network_name)
# show results
formattable(head(all_nws))| network_name | species1 | species2 | connection_strength |
|---|---|---|---|
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus euxinicus | 3 |
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus hypanis | 3 |
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus proximus | 74 |
| A_HP_001 | Apodemus mystacinus | Ctenophthalmus shovi | 13 |
| A_HP_001 | Apodemus mystacinus | Hystrichopsylla satunini | 4 |
| A_HP_001 | Apodemus mystacinus | Leptopsylla segnis | 11 |
Another useful functionality provided by the dplyr package is the pipe operator %>% which allows passing the output of a function to another one. For instance,
formattable(head(all_nws))
# is equivalent to
head(all_nws) %>% formattable()Networks can be filtered by interaction type passing this as an argument in the url. Accepted values of the parameter interaction_type are:
* Anemone-Fish
* FoodWebs
* Host-Parasite
* Plant-Ant
* Plant-Herbivore
* Pollination
* SeedDispersal
* [default ALL]
Here the example for pollination networks:
json_url <- paste0(base_url,"get_networks.php?interaction_type=Pollination")
pol_nws <- jsonlite::fromJSON(json_url)Networks can also be filtered by data type setting the parameter data_type equals to weighted or binary [default ALL]. To download weighted, pollination networks you can run the command:
json_url <- paste0(base_url,"get_networks.php?interaction_type=Pollination&data_type=weighted")
pol_weighted_nws <- jsonlite::fromJSON(json_url)1.1.3 Download network metadata from the weboflife API
You can obtain a csv file containing the information about species in a given network hitting the endpoint https://www.web-of-life.es/get_species_info.php and passing the network name into the url, namely
# define the base url
base_url <- "https://www.web-of-life.es/"
# download dataframe
M_PL_073_info <- read.csv(paste0(base_url,"get_species_info.php?network_name=M_PL_073"))
# show results
head(M_PL_073_info) # %>% formattable()## species abundance role is.resource taxonomic.resolution
## 1 Calystegia sepium 4 Plant 1 Species
## 2 Lapsana communis 17 Plant 1 Species
## 3 Polygonum amphibium 10 Plant 1 Species
## 4 Apocynum cannabinum 10 Plant 1 Species
## 5 Asclepias syriaca 58 Plant 1 Species
## 6 Cephalanthus occidentalis 79 Plant 1 Species
## kingdom order family
## 1 Plants Solanales Convolvulaceae
## 2 Plants Asterales Asteraceae
## 3 Plants Caryophyllales Polygonaceae
## 4 Plants Gentianales Apocynaceae
## 5 Plants Gentianales Apocynaceae
## 6 Plants Gentianales RubiaceaeYou can obtain a csv file with the metadata of all the networks hitting the endpoint
https://www.web-of-life.es/get_network_info.php:
all_nw_info <- read.csv(paste0(base_url,"get_network_info.php"))
# see what information is provided
colnames(all_nw_info)## [1] "network_name" "network_type"
## [3] "author" "reference"
## [5] "number_components" "is_weighted"
## [7] "cell_values_description" "abundance_description"
## [9] "location" "location_address"
## [11] "country" "latitude"
## [13] "longitude"# select only some specific columns
my_nw_info <- select(all_nw_info, network_name, location, latitude, longitude)
head(my_nw_info) %>% formattable()| network_name | location | latitude | longitude |
|---|---|---|---|
| M_PL_027 | Arthur’s Pass, New Zealand | -42.95000 | 171.566667 |
| M_PL_021 | Ashu, Kyoto, Japan | 35.33333 | 135.750000 |
| M_PL_017 | Bristol, England | 51.57499 | -2.589902 |
| M_PL_032 | Brownfield, Illinois, USA | 40.13333 | -88.166667 |
| M_SD_003 | Caguana, Puerto Rico | 18.29694 | -66.781944 |
| M_SD_015 | Campeche state, Mexico | 18.50698 | -89.486184 |
For completeness, we mention that passing the initial part of a network name into the url you can obtain the metadata for networks whose name starts, e.g., with M_PL_072
my_nws_info <- read.csv(paste0(base_url,"get_network_info.php?network_name=M_PL_072")) %>%
select(network_name, location, latitude,longitude)
# visualize data
my_nws_info %>% formattable()| network_name | location | latitude | longitude |
|---|---|---|---|
| M_PL_072 | Pampas, Argentina | -36.88333 | -63.03333 |
| M_PL_072_01 | Amarante, Pampas, Argentina | -37.84201 | -58.35857 |
| M_PL_072_02 | Cinco Cerros, Pampas, Argentina | -37.73903 | -58.23154 |
| M_PL_072_03 | Difuntito, Pampas, Argentina | -37.75721 | -58.24847 |
| M_PL_072_04 | Difuntos, Pampas, Argentina | -37.88535 | -57.84195 |
| M_PL_072_05 | El Morro, Pampas, Argentina | -37.73848 | -58.43347 |
| M_PL_072_06 | La Barrosa, Pampas, Argentina | -37.87348 | -58.26069 |
| M_PL_072_07 | La Brava, Pampas, Argentina | -37.87288 | -57.99225 |
| M_PL_072_08 | La Chata, Pampas, Argentina | -37.87546 | -58.38000 |
| M_PL_072_09 | La Paja, Pampas, Argentina | -37.75336 | -58.28484 |
| M_PL_072_10 | Piedra Alta, Pampas, Argentina | -37.73224 | -58.31179 |
| M_PL_072_11 | Vigilancia, Pampas, Argentina | -37.87268 | -58.03099 |
| M_PL_072_12 | Volcan, Pampas, Argentina | -37.85078 | -58.06569 |
1.1.4 Exercise 1
- How many networks are stored in the Web of life databases? how many pollination and seed-dispersal networks?
- How many entries in the M_PL_073 network are not taxonomically resolved down to the species level?
- How many networks in Web of life have been collected in the tropical region?
Hint: to filter w.r.t. to a latitude between the two tropics one can use function filter() of the dplyr package for data manipulation:
tropical_nws_info <- my_nws_info %>% filter(.,between(latitude, -23.43647, 23.43647)) 1.2 Analyze networks with igraph
In the scientific literature the words network and graph are used interchangeably, which typically confuses students who approach this topic. It is therefore convenient to keep in mind the following correspondence table
| Graph Theory | Network Science |
|---|---|
| graph | network |
| vertex | node |
| edge | link |
Graph Theory is a traditional branch of mathematics, which started during the 18th century. Network Science refers more (but not only!) to real systems; as such the latter exploded at the beginning of this century, as more and more data became available (www, internet, cell-phone or social-media contacts, etc.). In an ecological network nodes represent species and links their pairwise interactions.
The reference package for graph/network analysis is igraph.
It is an open source software, optimized by professional developers and can be invoked from R, Python, Mathematica, and C/C++. It contains a lot of libraries, though you will explore only few of them throughout this course.
The recommendation is that, whenever you need to do some operation or calculation with networks, you first look if that functionality is already provided in igraph. If not, you can probably build on some existing functions to develop your own function/package.
Though properly specking graph refers to a general mathematical entity, in this document we find it convenient to indicate with the word graph an igraph object in the context of the igraph package. Network data can be represented in several ways, corresponding to different data structures (classes). Below we will consider the following ones:
| representation | data structure |
|---|---|
| edge list | dataframe |
| graph | igraph object |
| adjacency matrix | matrix/array |
| incidence matrix | matrix/array |
1.2.1 From dataframe to igraph object
The network data downloaded from Web of life are stored in a representation (network_name, species1, species2, connection_strength). The last three columns correspond to the edgelist representation of a graph. Therefore, the values contained in those three columns can be converted into a graph object using the function graph_from_data_frame() of the igraph package
# download the foodweb FW_001
json_url <- paste0(base_url,"get_networks.php?network_name=FW_001")
FW_001_nw <- jsonlite::fromJSON(json_url)
# check the class
class(FW_001_nw) ## [1] "data.frame"# import the igraph package (if not done at the beginning)
library(igraph)
# select the 3 relevant columns and create the igraph object
FW_001_graph <- FW_001_nw %>% select(species1, species2, connection_strength) %>%
graph_from_data_frame(directed = TRUE)
# check the class
class(FW_001_graph) ## [1] "igraph"As you can see from the last command, we have just created an igraph object. Note that, since in foodwebs it is relevant who eats whom, the representative graph is directed. This is specified setting the corresponding parameter as TRUE in the function graph_from_data_frame().
1.2.2 Plot a foodweb as a directed graph
Once the network is represented as an igraph object it can easily be visualized
plot(FW_001_graph, vertex.size=4,
vertex.label=NA,
edge.arrow.size= 0.3,
layout = layout_on_sphere) 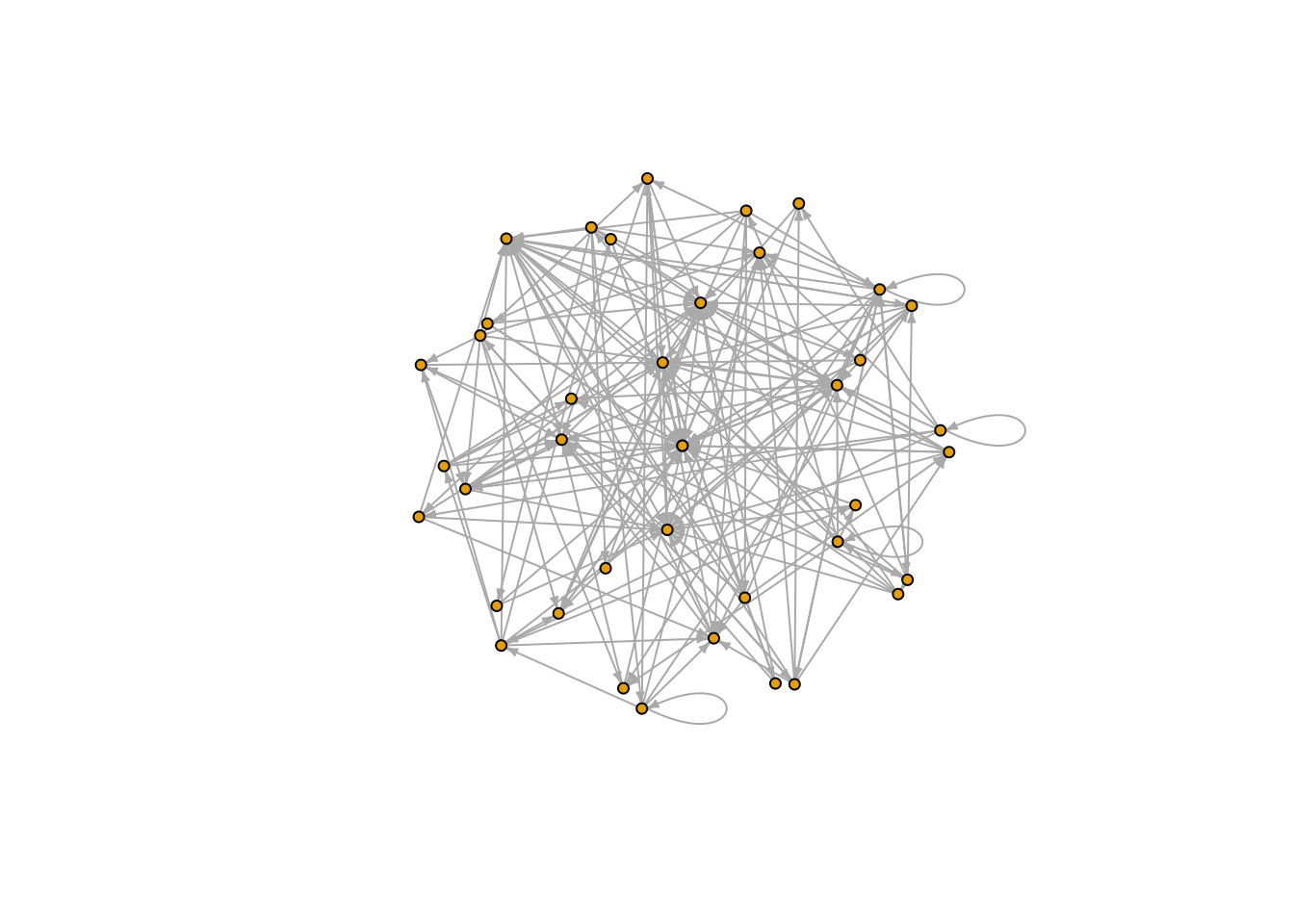
Other possible layouts are: layout_nicely, layout_with_kk, layout_randomly, layout_in_circle, layout_on_sphere.
1.2.3 From igraph object to adjacency matrix
Another way to represent an ecological network is the adjacency matrix, that is a square matrix whose rows and columns run over all the species name in the network and whose elements indicate the strength of interaction between species pairs (zero means no interaction). This data structure is generally less efficient from the perspective of memory storage w.r.t. an edge list, but it can be useful to facilitate visualization or to perform some calculations (e.g., evaluate the nestedness of a network, perform spectral analysis, etc.).
Using the function as_adjacency_matrix() of the igraph package
the adjacency matrix associated with a given network can be created. Let us focus on the relatively small FW_002 foodweb. Some care must be put in passing the right arguments to the function as_adjacency_matrix()
because the graph is directed. The reader is addressed to the
user manual for more details.
# download the foodweb FW_002
json_url <- paste0(base_url,"get_networks.php?network_name=FW_002")
FW_002_nw <- jsonlite::fromJSON(json_url)
# select the 3 relevant column and pass and create the igraph object
FW_002_graph <- FW_002_nw %>% select(species1, species2, connection_strength) %>%
graph_from_data_frame(directed = TRUE)
# convert the igraph object into adjacency matrix
adj_matrix <- as_adjacency_matrix(FW_002_graph,
type = "lower", # it is directed!!!
attr = "connection_strength",
sparse = FALSE) If you give a quick look at the variable adj_matrix running the command
head(adj_matrix)you will notice that it does not produce exactly what we want: 1. elements are strings, instead of numerical values, and 2. there are some empty strings to indicate that a pair of species do not interact. We can fix both problems as follows
# convert elements into numeric values
class(adj_matrix) <-"numeric"
# remove NA values (previous empty strings)
adj_matrix[which(is.na(adj_matrix) == T)]<-0
# check dimensions and compare with the number of species on the Web interface
dim(adj_matrix)## [1] 14 14# convert the adjacency matrix into a dataframe just for visualization purposes
df <- adj_matrix %>% as.data.frame()
df <- df[order((rownames(df))),] # to order by column names
# visualize the adjacency matrix
df %>% formattable()| Leporinus friderici | Astyanax altiparanae | Hoplias malabaricus | Detritus | Phytoplankton | Macrophytes | Terrestrial Invertebrates | Astyanax fasciatus | Brycon nattereri | Hypostomus strigaticeps | Detritus Animal | Import | Salminus hilarii | Zooplankton | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Astyanax altiparanae | 0.000 | 0.0000 | 0.500 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.740 | 0.0 |
| Astyanax fasciatus | 0.000 | 0.0000 | 0.100 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.060 | 0.0 |
| Brycon nattereri | 0.000 | 0.0000 | 0.000 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.115 | 0.0 |
| Detritus | 0.000 | 0.0000 | 0.001 | 0 | 0 | 0 | 0.3 | 0.000 | 0.003 | 1 | 0 | 0 | 0.000 | 0.6 |
| Detritus Animal | 0.010 | 0.0006 | 0.001 | 0 | 0 | 0 | 0.0 | 0.307 | 0.001 | 0 | 0 | 0 | 0.000 | 0.0 |
| Hoplias malabaricus | 0.000 | 0.0000 | 0.030 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.000 | 0.0 |
| Hypostomus strigaticeps | 0.000 | 0.0000 | 0.000 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.010 | 0.0 |
| Import | 0.252 | 0.0290 | 0.000 | 0 | 0 | 0 | 0.5 | 0.000 | 0.219 | 0 | 0 | 0 | 0.000 | 0.0 |
| Leporinus friderici | 0.000 | 0.0000 | 0.000 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.050 | 0.0 |
| Macrophytes | 0.100 | 0.1000 | 0.007 | 0 | 0 | 0 | 0.2 | 0.500 | 0.036 | 0 | 0 | 0 | 0.000 | 0.0 |
| Phytoplankton | 0.004 | 0.0200 | 0.000 | 0 | 0 | 0 | 0.0 | 0.001 | 0.000 | 0 | 0 | 0 | 0.000 | 0.4 |
| Salminus hilarii | 0.000 | 0.0000 | 0.000 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.000 | 0.0 |
| Terrestrial Invertebrates | 0.634 | 0.8500 | 0.361 | 0 | 0 | 0 | 0.0 | 0.192 | 0.742 | 0 | 0 | 0 | 0.025 | 0.0 |
| Zooplankton | 0.000 | 0.0000 | 0.000 | 0 | 0 | 0 | 0.0 | 0.000 | 0.000 | 0 | 0 | 0 | 0.000 | 0.0 |
1.2.4 Bipartite networks
As you will learn in the classes, several ecological networks are bipartite:
1. interactions are not directed
2. species can be split in two groups and interactions occurs only from one group to the other.
Typical examples are pollination or seed-dispersal networks. Let us see how these features of bipartite networks affect the way in which their data has to be handled with igraph.
We first download the seed-dispersal network M_SD_002 and convert it into an igraph object:
# download the foodweb M_SD_002
json_url <- paste0(base_url,"get_networks.php?network_name=M_SD_002")
M_SD_002_nw <- jsonlite::fromJSON(json_url)
# select the 3 relevant columns and create the igraph object
M_SD_002_graph <- M_SD_002_nw %>% select(species1, species2, connection_strength) %>%
graph_from_data_frame() Before plotting this graph or representing it as a bipartite incidence matrix, one needs to determine which nodes of the graph M_SD_002_graph belong to one or the other group, e.g., plants or animals.
The function bipartite.mapping() does the job of splitting the nodes in two groups, if the criterion 2. is actually met.
bipartite.mapping(M_SD_002_graph)## $res
## [1] TRUE
##
## $type
## [1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE TRUE TRUE TRUE
## [13] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
## [25] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
## [37] TRUE TRUE TRUE TRUEWe can save this information into a vector isGroupA and pass it to the object M_SD_002_graph defining as TRUE/FALSE the type for each vertex in the graph; igraph uses this attribute of vertices to identify the two groups of species.
# Define the vector of booleans
isGroupA <- bipartite_mapping(M_SD_002_graph)$type
# Add the "type" attribute to the vertices of the graph
V(M_SD_002_graph)$type <- isGroupA The way in which igraph assigns species (nodes) to one the other group is arbitrary. However, we would like to associate one guild to resources and the other to consumers. The two group of species will then be displayed in the incidence matrix systematically as rows and columns, respectively. This information can also be retrieved from Web of life:
# get info about species
my_info <- read.csv(paste0(base_url,"get_species_info.php?network_name=M_SD_002"))
isResource <- my_info$is.resource %>% as.logical() # 0/1 converted to FALSE/TRUE
# Add the "type" attribute to the vertices of the graph
V(M_SD_002_graph)$type <- !(isResource) Now we can check that the igraph object was successfully converted into a bipartite graph running the following command
# check if igraph understands this object as bipartite
is_bipartite(M_SD_002_graph)## [1] TRUEIt is possible to pass from the representation of a bipartite graph to that of
incidence_matrix.
The latter is a matrix whose rows represent one group of species (e.g. plants) and columns the other group (e.g. animals).
This can be achieved using the function as_incidence_matrix() of the
igraph package.
Mutatis mutandis we first repeat the same steps done for the conversion from an igraph object to adjacency matrix
# convert the igraph object into incidence matrix
inc_matrix <- as_incidence_matrix(
M_SD_002_graph,
attr = "connection_strength",
names = TRUE,
sparse = FALSE
)
# convert elements into numeric values
class(inc_matrix) <-"numeric"
# remove NA values (previous empty strings)
inc_matrix[which(is.na(inc_matrix) == T)]<-0
# check dimensions and compare with the number of species on the Web interface
dim(inc_matrix)## [1] 31 9Now we would like to check whether this incidence matrix is consistent with the one displayed on the web interface of Web of life for the network M_SD_002
# convert the incidence matrix into a dataframe just for visualization purposes
df <- inc_matrix %>% as.data.frame()
df <- df[order((rownames(df))),] # to order by column names
#visualize the adjacency matrix
df %>% formattable()| Manucodia keraudrenii | Diphyllodes magnificus | Parotia lawesii | Paradisaea raggiana | Lophorina superba | Paradisaea rudolphi | Manucodia chalybatus | Ptiloris magnificus | Epimachus albertisii | |
|---|---|---|---|---|---|---|---|---|---|
| Aglaia sp1 M_SD_002 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 |
| Aporusa sp1 M_SD_002 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Canthium sp1 M_SD_002 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chisocheton sp1 M_SD_002 | 7 | 13 | 10 | 13 | 23 | 3 | 1 | 16 | 5 |
| Cissus aristata | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Cissus hypoglauca | 13 | 4 | 2 | 5 | 1 | 3 | 0 | 1 | 1 |
| Dysoxylum sp1 M_SD_002 | 1 | 12 | 1 | 17 | 0 | 0 | 0 | 0 | 0 |
| Elmerrillia sp1 M_SD_002 | 8 | 12 | 2 | 2 | 2 | 0 | 1 | 0 | 1 |
| Endospermum sp1 M_SD_002 | 2 | 15 | 2 | 2 | 9 | 0 | 0 | 0 | 0 |
| Ficus 181 | 7 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 |
| Ficus 202 | 2 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Ficus 217 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ficus 246 | 4 | 2 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Ficus 275 | 40 | 5 | 1 | 13 | 2 | 4 | 8 | 0 | 0 |
| Ficus 371 | 6 | 0 | 4 | 3 | 4 | 7 | 1 | 0 | 1 |
| Ficus drupacea | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ficus gul | 155 | 8 | 12 | 45 | 1 | 1 | 25 | 0 | 0 |
| Ficus odoardi | 55 | 8 | 6 | 20 | 1 | 0 | 19 | 2 | 0 |
| Gastonia sp1 M_SD_002 | 17 | 58 | 22 | 24 | 0 | 3 | 0 | 7 | 0 |
| Glochidion sp1 M_SD_002 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Homalanthus sp1 M_SD_002 | 4 | 104 | 10 | 75 | 17 | 23 | 0 | 8 | 0 |
| Myristica sp1 M_SD_002 | 3 | 1 | 3 | 1 | 0 | 0 | 0 | 0 | 0 |
| Pandanus sp1 M_SD_002 | 0 | 5 | 0 | 2 | 2 | 1 | 0 | 0 | 0 |
| Piper sp1 M_SD_002 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Schefflera sp1 M_SD_002 | 0 | 2 | 47 | 0 | 0 | 10 | 0 | 1 | 0 |
| Sloanea aberrans | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Sloanea sogerensis | 2 | 4 | 2 | 1 | 2 | 0 | 0 | 0 | 0 |
| Sterculia sp1 M_SD_002 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Syzygium sp1 M_SD_002 | 8 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Uvaria sp1 M_SD_002 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Zingiberaceae sp1 M_SD_002 | 0 | 2 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
After having downloaded and converted it into an incidence matrix, you can visualize the network M_SD_002 using the function plotweb() of the
bipartite package:
# import the bipartite package (if not done at the beginning)
library(bipartite)
plotweb(inc_matrix)
The two guilds within this bipartite network are shown on different layers: seed dispersers (top) and plants (bottom). Several parameters can be passed to the function plotweb() to fine-tune the output.
plotweb(inc_matrix,
method="normal", text.rot=90,
bor.col.interaction="gray40",plot.axes=F, col.high="blue", labsize=1,
col.low="darkgreen", y.lim=c(-1,3), low.lablength=40, high.lablength=40)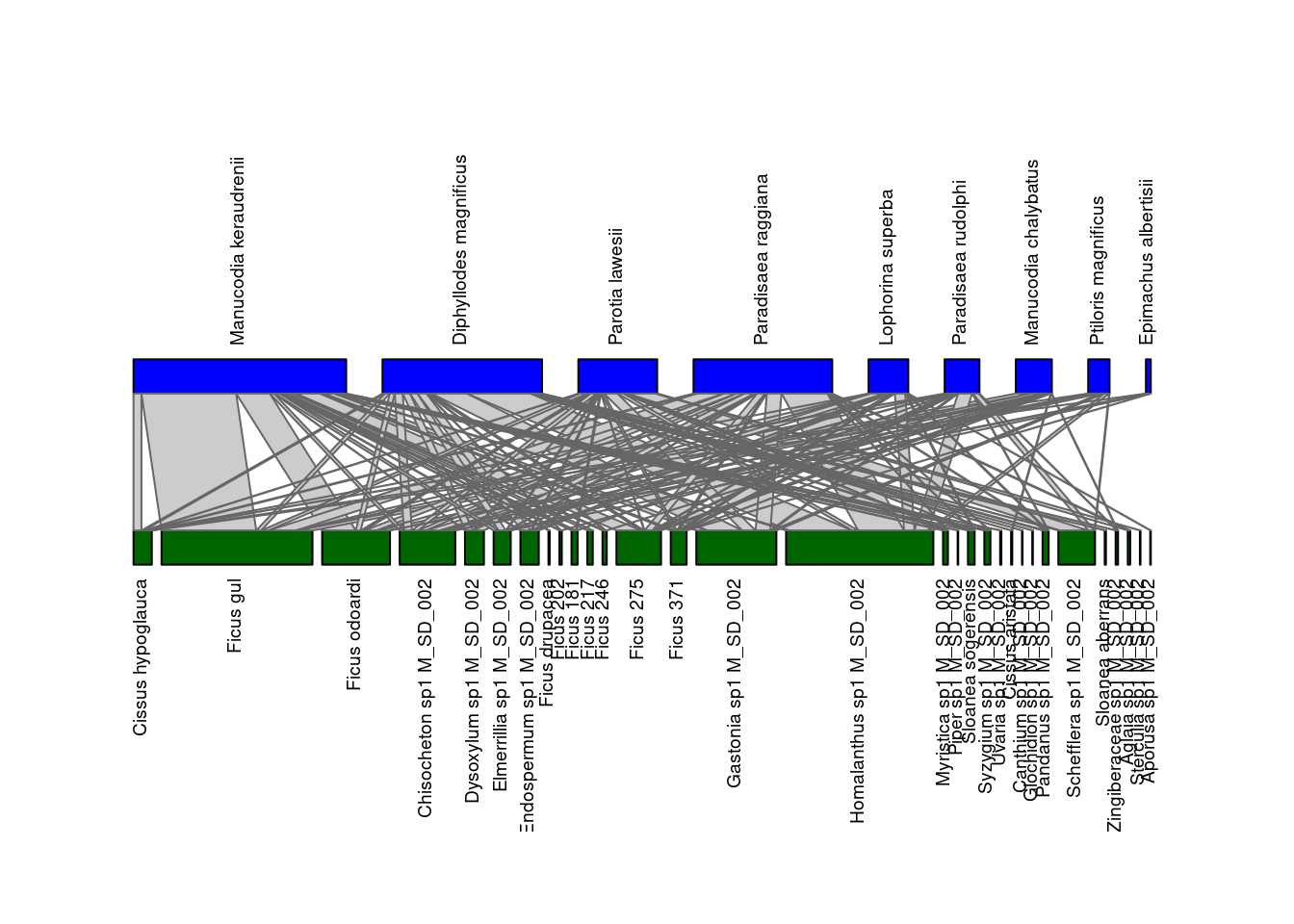
1.2.5 Exercise 2
From Web of life choose one foodweb on in the subset FW_017, download it, convert it into an adjacency matrix, and visualize it using igraph, using at least two layouts.
How is canibalism displayed in the plot of a directed graphs representing a foodweb?
Is the adjacency matrix corresponding to a foodwed symmetric?
Def.: Symmetrix is a matrix \(A\) for which \(A_{ij}=A_{ji}\).Download the pollination network M_PL_024 from Web of life, convert it into an incidence matrix, and visualize it using bipartite.
SAVE YOUR DATA: It is important that you save the data needed for a plot or analysis so that instructors can reproduce your output without running the whole script again. For this specific assignment, this can be done running the following command
save(FW_017_03_graph, adj_matrix,
M_PL_024_graph, inc_matrix,
file="~/ecological_networks_2023/workspace/03-16_toolkit_network_analysis/data_ex2.RData")- CHECK: The
*RDatafile should appear in the folder03-16_toolkit_network_analysis. If you want to be hundred percent sure that everything worked well, you can reload the variables from the created file:
load("~/ecological_networks_2023/workspace/03-16_toolkit_network_analysis/data_ex2.RData")and produce your plots again.
1.3 Connectance of pollination networks
The connectance is one of the properties characterizing a network and is defined as the ratio between the actual number of links (i.e. pairwise interactions among species) and the maximal number of links that could be realized in that type of network. For a bipartite network at most all the species of one group (e.g. plants) can interact with all the species of the other group (e.g. animals). Let us focus on pollination networks for which the connectance is defined as \[\begin{equation} \frac{\# \, links}{(\# \, plants) \times (\# animals)} \,. \end{equation}\] (The definition of connectance for a foodweb will be discussed in the questions and in forthcoming lectures).
1.3.1 Compute the connectance from an edge list
Our goal is to compute the connectance for all the pollination networks available on
Web of life
and store its value in a dataframe along with the size of the incidence matrix associated with each network.
We start downloading all the networks with interaction_type=Pollination
json_url <- paste0(base_url,"get_networks.php?interaction_type=Pollination")
pol_nws <- jsonlite::fromJSON(json_url)
head(pol_nws)## network_name species1 species2 connection_strength
## 1 M_PL_001 Centris cineraria Phacelia secunda 1
## 2 M_PL_001 Centris cineraria Senecio bustillosianus 1
## 3 M_PL_001 Centris cineraria Stachys albi 1
## 4 M_PL_001 Centris cineraria Adesmia conferta 1
## 5 M_PL_001 Centris cineraria Astragalus curvicaulis 1
## 6 M_PL_001 Centris cineraria Adesmia radicifolia 1When data, for a given network, is given in the form of edge list, computing the connectance is straightforward: one just needs to count the number of rows in the data frame (links) and the distinct rows in the column species1 (e.g. plants) and species2 (e.g. animals).
We first create a dataframe containing only the distinct network names
pol_nw_names <- distinct(pol_nws, network_name)Now we perform a for loop on the network names and compute the connectance and the size of the relative incidence matrix
# initialize dataframe to store results
pol_connectance_df <- NULL
for (nw_name in pol_nw_names$network_name){
nw <- filter(pol_nws, network_name == nw_name)
links <- nw %>% nrow()
plants <- distinct(nw, species2) %>% nrow()
animals <- distinct(nw, species1) %>% nrow()
size <- plants*animals
conn <- links/size
conn_row <- data.frame(nw_name, size, conn)
pol_connectance_df <- rbind(pol_connectance_df, conn_row)
}
colnames(pol_connectance_df) <- c("network_name", "size", "connectance")You can visualize the results as usual:
pol_connectance_df %>% head() %>% formattable() | network_name | size | connectance |
|---|---|---|
| M_PL_001 | 8484 | 0.04255068 |
| M_PL_002 | 2752 | 0.07122093 |
| M_PL_003 | 900 | 0.09000000 |
| M_PL_004 | 1224 | 0.13643791 |
| M_PL_005 | 26400 | 0.03496212 |
| M_PL_006 | 1037 | 0.14079074 |
# remove %>% head() to see the whole dataframe ACHTUNG: The Web of life API returns a dataframe in which only meaningful values for the connection_strength are given. Generally, it will be wise to check if all interaction values are non-vanishing and possibly filter out from the edge list the rows corresponding to vanishing or NA connection_strength. In pseudo code, this is done as follows
pol_nws_good <- filter(pol_nws, connection_strength > 0) %>%
filter(., !is.na(connection_strength)) The next plot shows the connectance of all pollination networks versus the incidence-matrix size for each network:
# import the ggplot2 package (if not done at the beginning)
library(ggplot2)
# define a function to plot as guide to the eye
guide_eye <- function(x,c,alpha){
return(c/x**alpha)
}
options(repr.plot.width=5, repr.plot.height=4)
ggplot() +
ggtitle("Pollination networks") +
stat_function(fun = function(x) guide_eye(x,4.8,0.5), color ="black", linetype = "dotted") +
geom_point(data = pol_connectance_df, aes(size, connectance), color = "purple", shape=1) +
labs(x = "Incidence matrix size",
y = "Network connectance") +
scale_x_continuous(trans='log10') +
scale_y_continuous(trans='log10') 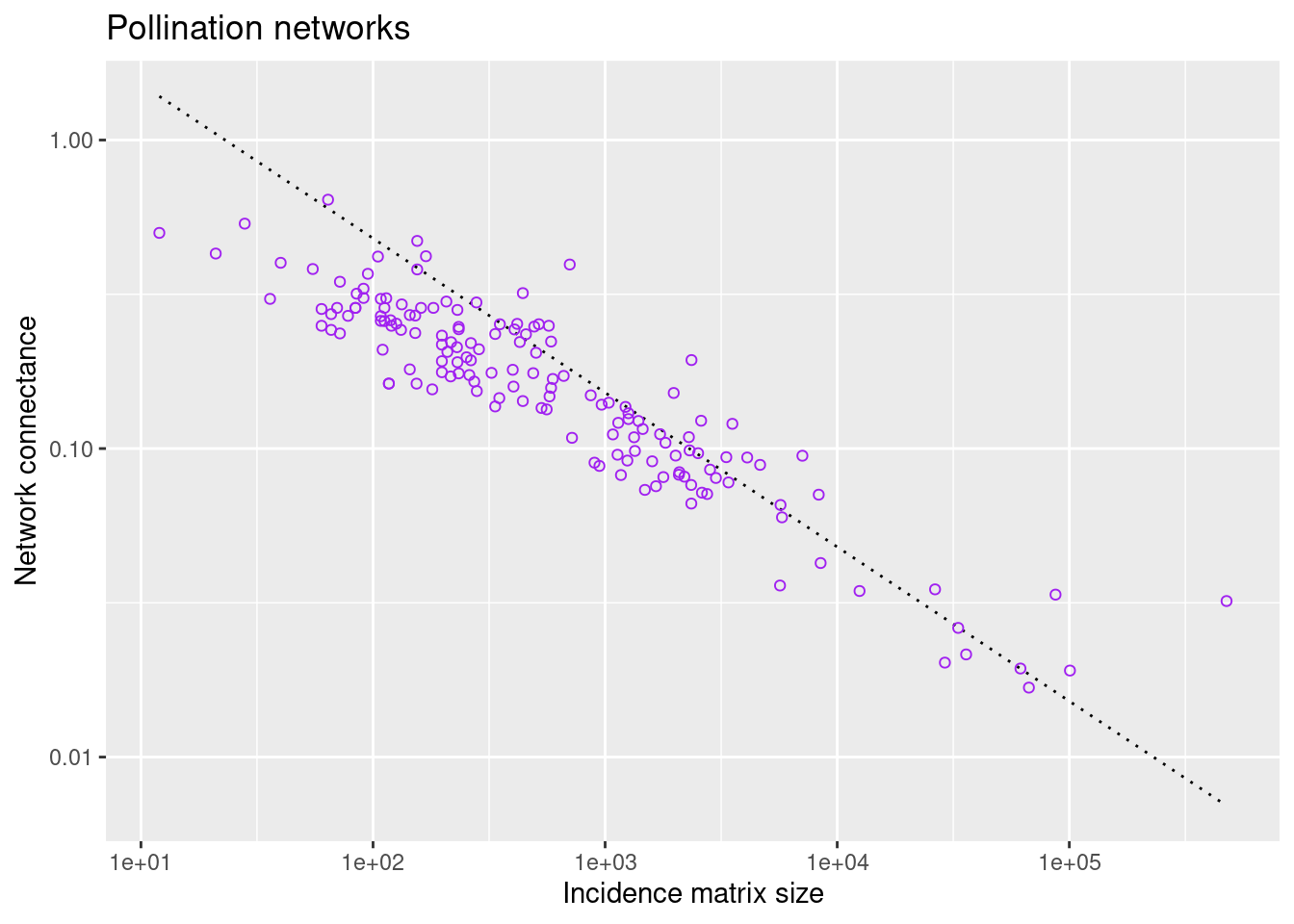
1.3.2 Filter by latitude
Now we would like to display with a different color the connectance of the subset of networks in the tropical area. We start downloading the metadata of all networks in Web of life (see above)
nws_info <- read.csv(paste0(base_url,"get_network_info.php"))
my_nws_info <- select(nws_info, network_name, location, latitude,longitude)From this dataframe we select the names of networks recorded in the tropical region, namely in the area with latitude between -23.43647\(^\circ\) and 23.43647\(^\circ\).
tropical_nws_info <- nws_info %>% filter(.,between(latitude, -23.43647, 23.43647))
tropical_nw_names <- distinct(tropical_nws_info, network_name) Now we keep in the dataframe pol_connectance_df only the data corresponding to tropical networks. In practice, we look for the intersection of the two ensembles: polination networks and tropical networks
tropical_pol_conn_df <- filter(pol_connectance_df, network_name %in% tropical_nw_names$network_name) %>%
select(network_name, size, connectance)At this point we can superimpose the (orange) points calculated for tropical networks to the previous plot
options(repr.plot.width=5, repr.plot.height=4)
ggplot() +
ggtitle("Tropical pollination networks") +
stat_function(fun = function(x) guide_eye(x,4.,0.5), color ="black", linetype = "dotted") +
geom_point(data = pol_connectance_df, aes(size, connectance), color = "purple", shape=1) +
geom_point(data = tropical_pol_conn_df, aes(size, connectance), color = "dark orange") +
labs(x = "Incidence matrix size",
y = "Network connectance") +
scale_x_continuous(trans='log10') +
scale_y_continuous(trans='log10') 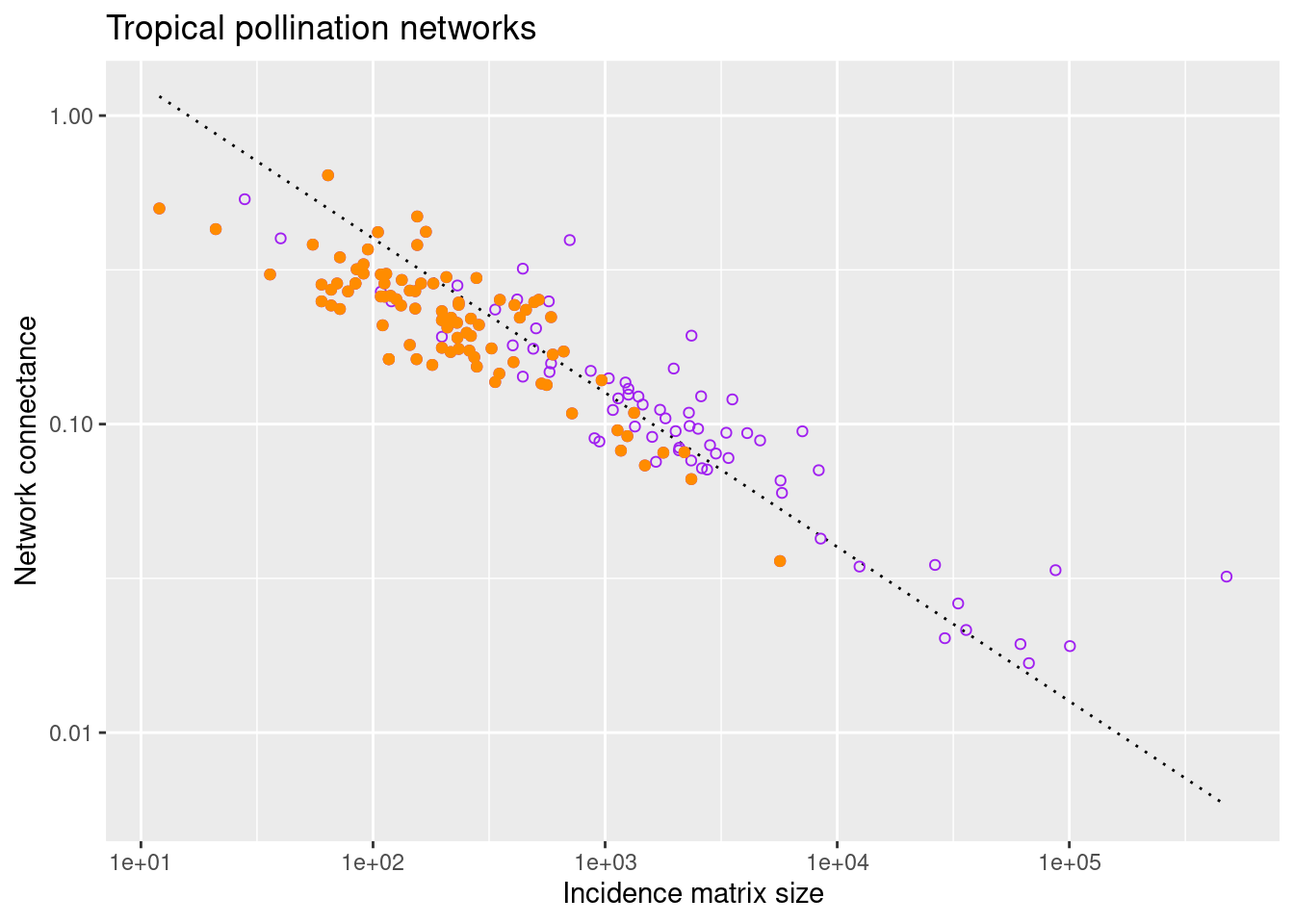
1.3.3 Exercise 3
Given that a food-web is typically represented by a square (adjacency) matrix, what would be in your opinion a sound definition of connectance in that case?
(Hint think of the maximal number of possible links and normalize accordingly)What is the functional dependence of the black line plotted on top of the graphs (note the scales are log-log)?
Superimpose the connectance computed for few selected networks at your choice to the previous plot and comment your observation.
(Hint insert the selected network names in the script provided below)
# put the selected network names in this vector as strings
my_nw_names <- c( )
my_pol_conn_df <- filter(pol_connectance_df, network_name %in% my_nw_names) %>%
select(network_name, size, connectance)
options(repr.plot.width=5, repr.plot.height=4)
ggplot() +
ggtitle("My pollination networks") +
stat_function(fun = function(x) guide_eye(x,4.,0.5), color ="black", linetype = "dotted") +
geom_point(data = pol_connectance_df, aes(size, connectance), color = "purple", shape=1) +
geom_point(data = my_pol_conn_df, aes(size, connectance), color = "black") +
# geom_point(data = tropical_pol_conn_df, aes(size, connectance), color = "dark orange") +
labs(x = "Incidence matrix size",
y = "Network connectance") +
scale_x_continuous(trans='log10') +
scale_y_continuous(trans='log10') - SAVE YOUR DATA: For this specific assignment, this can be save the relevant variables running the following command
save(pol_connectance_df, tropical_pol_conn_df, my_pol_conn_df,
file="~/ecological_networks_2023/workspace/03-16_toolkit_network_analysis/data_ex3.RData")- CHECK: Another
*RDatafile should appear in the folder03-16_toolkit_network_analysis. If you want to be hundred percent sure that everything worked well, you can reload the variables from the created file:
load("~/ecological_networks_2023/workspace/03-16_toolkit_network_analysis/data_ex3.RData")and produce your plots again.